Classical PCA
In classical PCA, given an input matrix $\mathbf{X} \in \mathbb{R}^{N \times M}$ (In genetic settings, $N$ can be the number of individuals, and $M$ be the number of SNPs), the goal is to find an orthonormal matrix $W \in \mathbb{R}^{m\times K}$ containing $K$ orthonormal vectors; and the corresponding scores (weight) along each vector $z \in \mathbb{R}^K$ , such that each individual $\mathbf{X}_i \in \mathbb{R}^m$ can be reconstructed using $\hat{\mathbf{x}}_i = \mathbf{Wz}_i$ with the minimum error. Mathematically, we are trying the minimize: \(\begin{align} J(\mathbf{W},\mathbf{Z}) = \frac{1}{N} || \mathbf{X} - \mathbf{ZW}^T ||_F^2 \end{align}\)
Under the constraint that $\mathbf{W}$ is an orthonormal matrix, the PC scores for each individual are therefore uncorrelated. Moreover, it can be proved by induction that the orthonormal matrix $\mathbf{W}$ is the eigenvectors corresponding to the top-$K$ largest eigenvalues, which implies it captures the top-$K$ variance when projecting X into the lower dimensional orthonormal subspace.
EM PCA
If we treat the dimensional reduction method as a probabilistic model, then the score matrix $\mathbf{Z}$ becomes a probabilistic distribution, and suppose we have the following assumptions:
- Underlying latent variable has a Gaussian distribution
- There is a linear relationship between latent and observed variables
- Isotropic Gaussian noise (covariance proportional to an identity matrix) in observed dimension
Then we can set up the model as
\[\begin{align} \mathbf{x} = \mathbf{Wz} + \mathbf{\mu} + \mathbf{\epsilon} \\ P(\mathbf{z}) = \mathcal{N}(\mathbf{\mu}_0,\mathbf{\Sigma}) \\ P(\mathbf{x} | \mathbf{z}) = \mathcal{N}(\mathbf{Wz} + \mathbf{\mu}_0, \sigma^2\mathbf{I}) \end{align}\]Notice here we can assume $\mathbf{\mu}_0 = \pmb{0}$ and $\mathbf{\Sigma} = \mathbf{I}$ without losing generality, since if they are not, we can always find another $\mathbf{W}’ = \mathbf{WU}$ such that $\mathbf{Uz} \sim \mathcal{N}(\pmb{0},\mathbf{I})$.
Then the marginal probability of $\mathbf{x}$ can be expressed as \(\begin{align} p(\mathbf{x}) = \mathcal{N}(\mathbf{\mu},\mathbf{WW}^T+\sigma^2\mathbf{I}) \end{align}\)
To see this, notice that \(\begin{align} \mathbb{E}[\mathbf{x}] = \mathbb{E}[\mathbf{\mu+Wz+\epsilon}] = \mathbf{\mu} + \mathbf{W}\mathbb{E}[\mathbf{z}] + \mathbb{E}[\mathbf{\epsilon}] = \mu \\ Var(\mathbb{x}) = \mathbb{E}[(\mathbf{\mu+Wz+\epsilon})(\mathbf{\mu+Wz+\epsilon})^T] = \mathbf{WW}^T + \sigma^2\mathbf{I} \end{align}\)
Further, the covariance can be easily calculated as \(\begin{align} Cov[\mathbf{x},\mathbf{z}] & = \mathbb{E}[(\mathbf{x}-\mathbf{\mu})(\mathbf{z}-\mathbf{0})^T] \\ &= \mathbb{E}[\mathbf{xz}^T - \mathbf{\mu z}^T] \\ &= \mathbb{E}[\mathbf{(Wz + \mu + \epsilon)z}^T] - \mathbf{\mu}\mathbb{E}[\mathbf{z}^T] \\ &= \mathbf{W}\mathbb{E}[\mathbf{zz}^T] \\ & = \mathbf{W} \end{align}\)
Then the joint probability is: \(p\left(\begin{bmatrix} \mathbf{z} \\ \mathbf{x} \end{bmatrix}\right) = \mathcal{N}\left(\begin{bmatrix} \mathbf{z} \\ \mathbf{x} \end{bmatrix} \bigg| \begin{bmatrix} \mathbf{0} \\ \boldsymbol{\mu} \end{bmatrix}, \begin{bmatrix} \mathbf{I} & \mathbf{W}^T \\ \mathbf{W} & \mathbf{WW}^T + \sigma^2\mathbf{I} \end{bmatrix}\right)\)
Applying Gaussian conditional probability, we get: \(p(\mathbf{z|x}) = \mathcal{N}(\mathbf{z | m, V}), \quad \mathbf{m} = \mathbf{W}^T(\mathbf{WW}^T + \sigma^2\mathbf{I})^{-1}(\mathbf{x} - \boldsymbol{\mu}), \quad \mathbf{V} = \mathbf{I} - \mathbf{W}^T(\mathbf{WW}^T + \sigma^2\mathbf{I})^{-1}\mathbf{W}\)
We can simplify the problem by standardizing our dataset $\pmb{X}$ so that $\pmb{\mu} = 0$. We, therefore, complete the setup of a typical EM algorithm.
In E-step, we compute \(% \begin{align} \lim_{\sigma \to 0}p(\mathbf{Z}|\mathbf{X}) = \mathbf{W}^T(\mathbf{WW}^T)^{-1}\mathbf{X}^T % \end{align}\) If $\sigma \neq 0$, the results would become probabilistic, in which case we don’t discuss. Notice that $\pmb{WW}^T$ is an $m \times m$ matrix, which takes $O(m^3)$ to compute the inverse. We instead cleverly apply the matrix inverse property to transform $\pmb{W}^T(\pmb{WW}^T)^{-1}$ to $(\pmb{W}^T\pmb{W})^{-1}\pmb{W}^T$, which reduces the inverse computation to $O(K^3)$, so that \(% \begin{align} \pmb{\hat{Z}} = (\pmb{W}^T\pmb{W})^{-1}\pmb{W}^T\pmb{X}^T % \end{align}\) In M-step, we compute the Q function \(\begin{align} & Q(\theta, \theta^{(t)}) = E[log(\pmb{X},\pmb{Z}| \pmb{W}, \sigma^2] \\ &= \sum_{i=1}^n p(\pmb{z}_i|\pmb{x}_i)(log(p(\pmb{x}_i|\pmb{z}_i)) + log(p(\pmb{z}_i))) \end{align}\) and by taking the partial derivative for $\pmb{W}$, we get \(% \begin{align} \pmb{\hat{W}} = \pmb{X}\pmb{Z}^T(\pmb{Z\pmb{Z}^T})^{-1} % \end{align}\) We thus complete the construction of the EM-PCA algorithm. Notice the complexity of the EM-PCA algorithm is dominated by $O(TKmn)$, and T is the number of iterations. This algorithm is linear regarding sample size and feature dimension, therefore bringing great advantage when the reduction dimension $K \ll m$ and $K \ll n$.
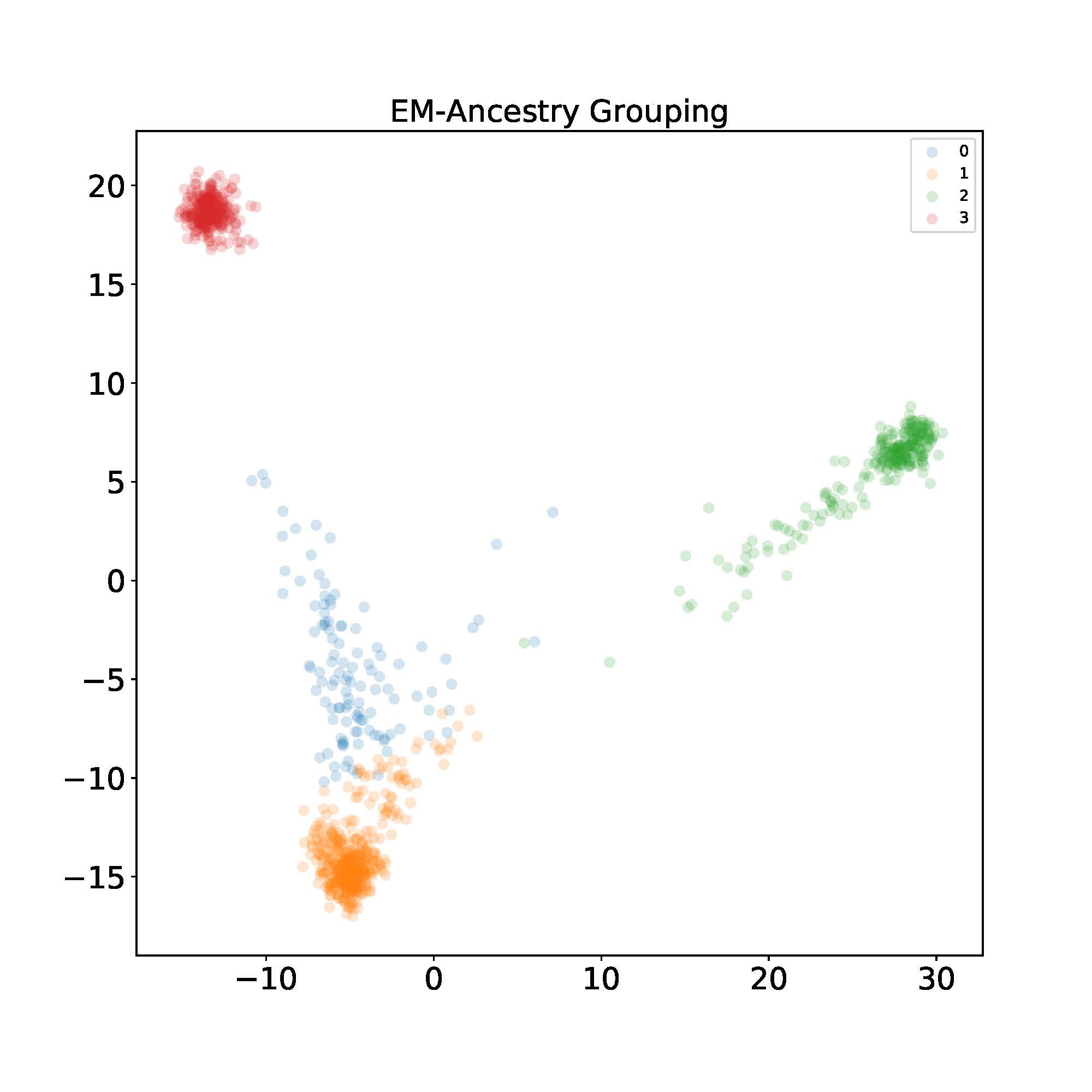
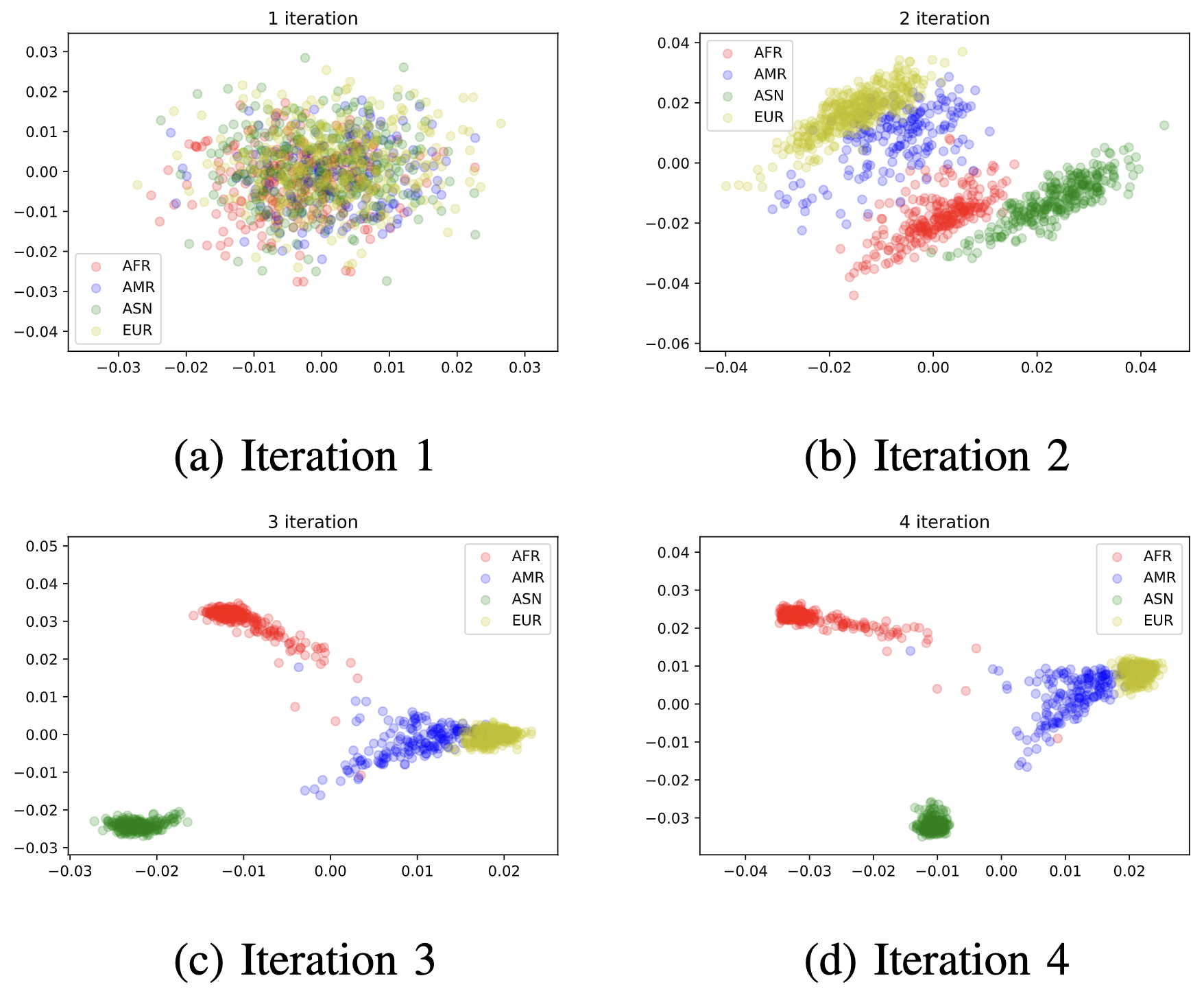
Experimental Results
Now let’s apply the algorithm to the 1000 Genome dataset to see how well the algorithm performs. As can be easily seen, EM-PCA has a fast convergence rate with a small runtime complexity.
References:
- Murphy, Kevin P. Machine learning: a probabilistic perspective. MIT press, 2012.
- Siva, Nayanah. “1000 Genomes project.” Nature biotechnology 26.3 (2008): 256-257.